Download A4Medicine Mobile App
Empower Your RCGP AKT Journey: Master the MCQs with Us!
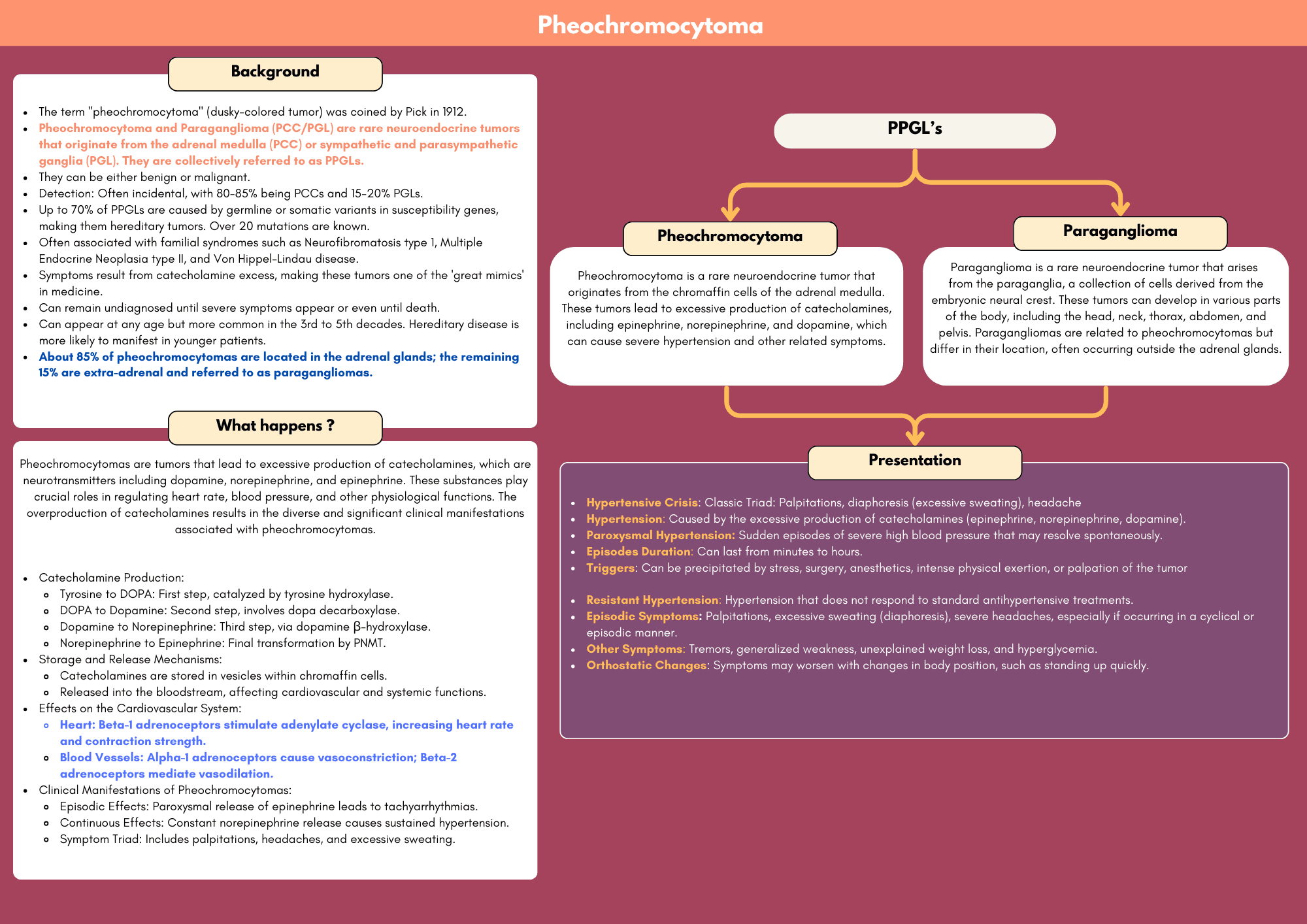
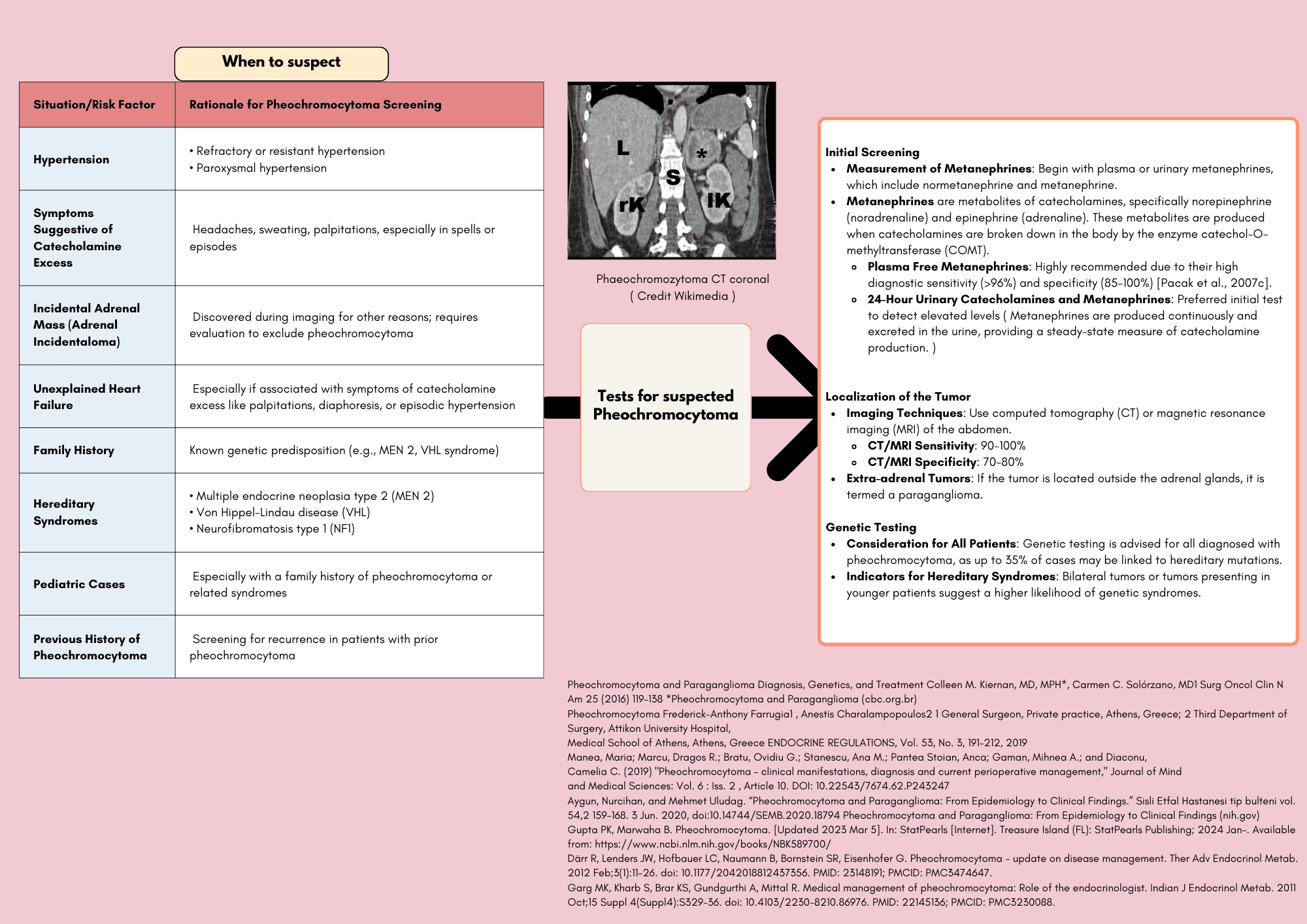
Pheochromocytoma is a rare neuroendocrine tumor originating from chromaffin cells in the adrenal medulla, leading to excessive production of catecholamines, primarily adrenaline and noradrenaline. This condition is a critical consideration for clinicians due to its potentially life-threatening cardiovascular effects, including hypertension, tachycardia, and arrhythmias.
The presentation of pheochromocytoma can vary, often characterized by episodic symptoms such as severe headaches, palpitations, diaphoresis, and paroxysmal hypertension. These symptoms can mimic other conditions, making diagnosis challenging. However, the classic triad of headaches, sweating, and tachycardia, alongside elevated plasma and urinary catecholamine levels, can be key indicators.
Pheochromocytomas can be sporadic or associated with genetic syndromes such as Multiple Endocrine Neoplasia type 2 (MEN2), Von Hippel-Lindau disease, and Neurofibromatosis type 1 (NF1). Comprehensive evaluation including biochemical testing, imaging studies, and genetic counseling is essential for accurate diagnosis and management.
The definitive treatment for pheochromocytoma is surgical resection, which requires meticulous preoperative management to control hypertension and prevent intraoperative catecholamine surges. Long-term follow-up is crucial to monitor for recurrence or metastasis.
Baseline Primary Care Investigations for Suspected Pheochromocytoma
| Investigation | Purpose | Description |
|---|---|---|
| Clinical History and Physical Examination | Assess symptoms and identify risk factors | Look for symptoms such as hypertension, palpitations, headaches, and sweating. Check for family... |
Try our Free Plan to get the full article.
